

巨噬细胞凋亡对动脉粥样硬化斑块的发展
发布日期:2023-08-15 浏览次数:1463

背景
动脉粥样硬化是世界上死亡的主要原因。研究证实巨噬细胞凋亡对动脉粥样硬化斑块的发展至关重要。巨噬细胞识别并摄取氧化低密度脂蛋白(ox-LDL),然后转化为泡沫细胞,发生凋亡,最终促进斑块不稳定性。
自噬是一种与凋亡密切相关的自我保护机制,它依赖于溶酶体降解和循环细胞内成分来维持体内稳态。位点自噬流是指自噬的整个动态过程,包括自噬体的形成、自噬体与溶酶体的融合以及自噬体的降解。超过30个自噬相关基因(Atgs)和蛋白参与了自噬过程。LC3I向LC3II的转化是自噬激活的标志,而p62的降解被认为是自噬蛋白降解的标志,p62蛋白的表达量与自噬活性呈现负相关。巨噬细胞自噬通过增加胆固醇流出在动脉粥样硬化中起保护作用。此外,在Ldlr -/-小鼠中,巨噬细胞Atg5缺乏会增加细胞凋亡并促进斑块坏死。因此,基于细胞凋亡和自噬调节的治疗策略可能适合动脉粥样硬化。
自噬的调控是复杂的,涉及多种途径。声动力疗法通过诱导ROS产生促进巨噬细胞自噬。此外,肌磷脂3激酶(PI3K)/蛋白激酶B (AKT)/雷帕霉素(mTOR)是一个经典的自噬途径。同时,激活ERK1/2也可诱导自噬。转录因子EB (TFEB)是mTOR和ERK1/2的下游靶点,是自噬-溶酶体通路的关键调控因子。TFEB上调近三分之二的自噬-溶酶体途径相关基因的表达,其过表达通过挽救脂质诱导的溶酶体功能障碍和下游后遗症,在动脉粥样硬化中具有强大的治疗作用。
诱导自噬和抑制细胞凋亡可能为动脉粥样硬化(AS)提供一种治疗方法,巨噬细胞自噬的调控被证明对动脉粥样硬化非常有益。然而,很少有自噬调节剂的报道。ATO通过促进肿瘤细胞凋亡和自噬死亡在急性早幼粒细胞白血病中发挥治疗作用。提示,ATO可调节巨噬细胞的凋亡和自噬,为动脉粥样硬化的治疗提供了新的视角。
1. 三氧化二砷(ATO)诱导巨噬细胞自噬
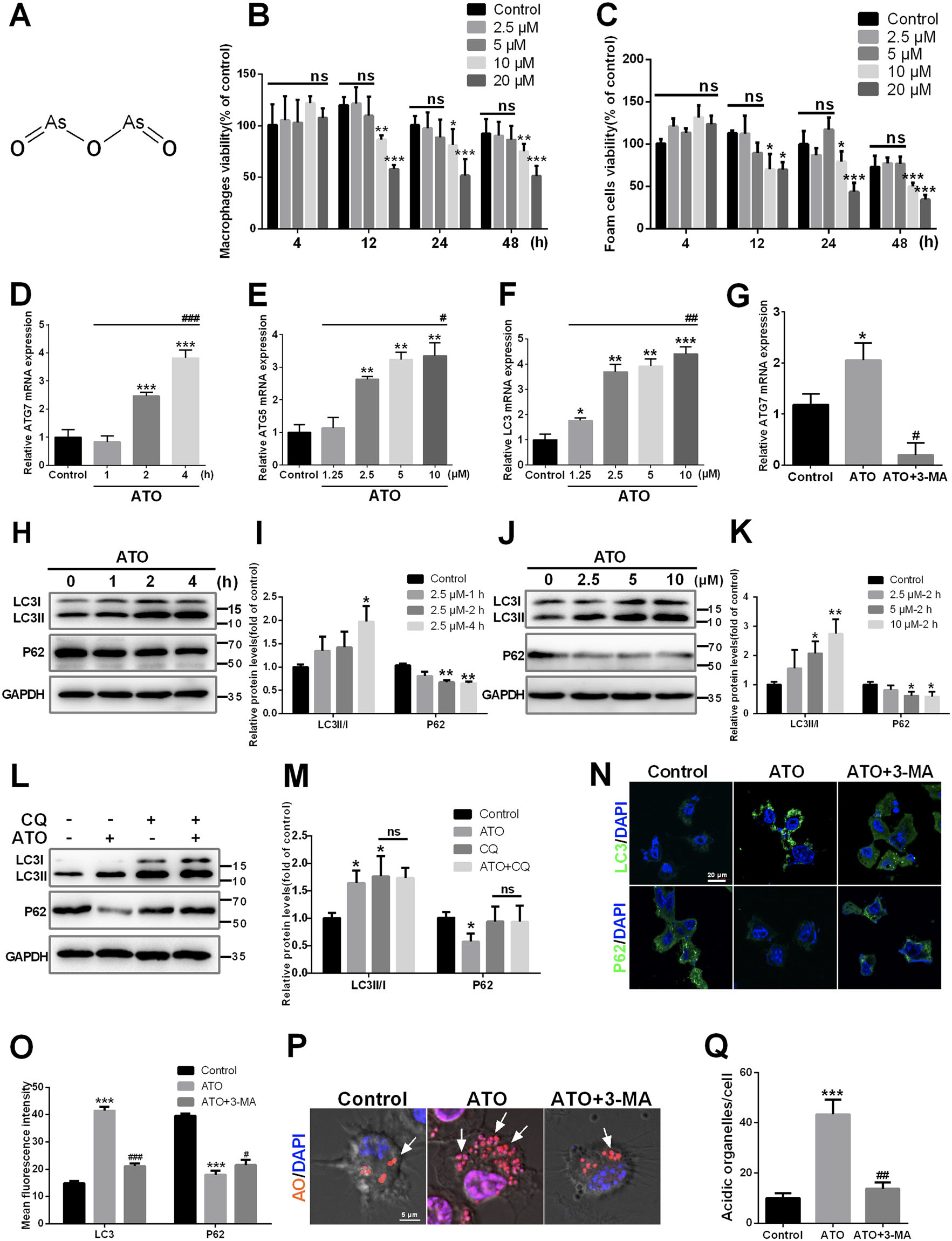
MTT实验证明ATO对巨噬细胞和泡沫细胞活力影响具有时间和剂量依赖性
qPCR:ATO处理促进ATG7、ATG5、LC3的表达
WB:ATO增加LC3II/LC3I比值,抑制P62的表达具有时间和剂量依赖性
免疫荧光:表明自噬抑制剂抑制了 ATO诱导的Atg7 mRNA表达、LC3II的积累和p62的降解。
吖啶橙(AO)染色:显示 ATO增加红色信号(酸性细胞器)。另一方面,
3-MA消除了ATO诱导的细胞器酸化
这些数据表明,ATO促进了自噬体的形成和溶酶体的自噬流。
2. 在巨噬细胞和泡沫细胞中,ATO诱导自噬的时间早于抑制凋亡标志物的表达
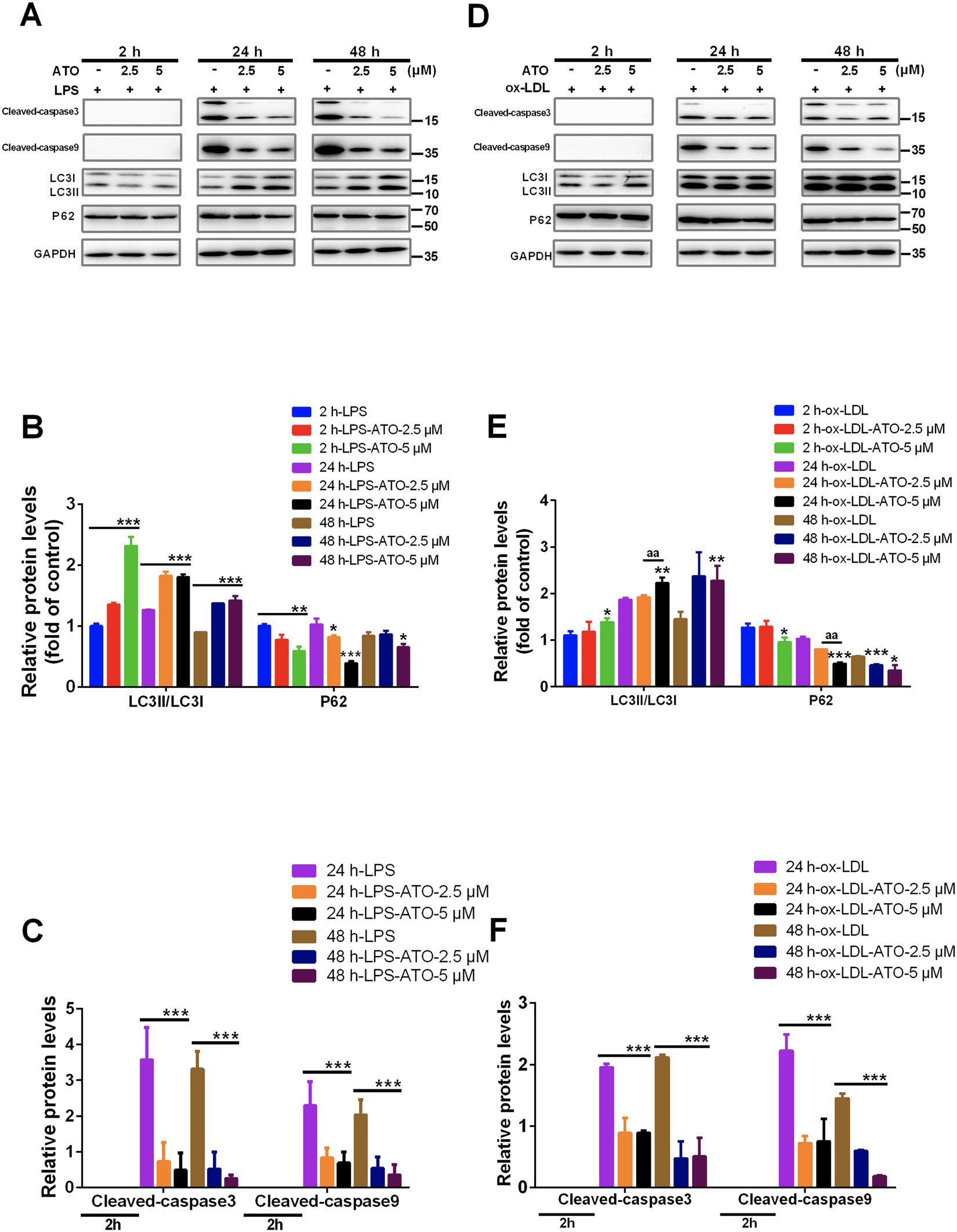
据报道,自噬和凋亡在不同的阶段被激活。Cleaved caspase-3和Cleaved caspase-9 是可靠的凋亡标志物
为了探讨ATO对自噬和凋亡的动态影响,检测了LC3II、p62、cl-caspase-3和cl-caspase-9的表达
表明 ATO 诱导巨噬细胞2h自噬,抑制细胞凋亡标志物24h、48h的表达
泡沫细胞中 ox-LDL 超载引起的细胞凋亡是一种动脉粥样硬化的迹象。模仿和探索动态ATO在此过程中对自噬和凋亡的影响
结果表明,ATO 2h时诱导泡沫细胞中的自噬,与其在 24 h时对细胞凋亡标志物的抑制相比要早得多。
3. ROS参与了巨噬细胞中ATO诱导的自噬
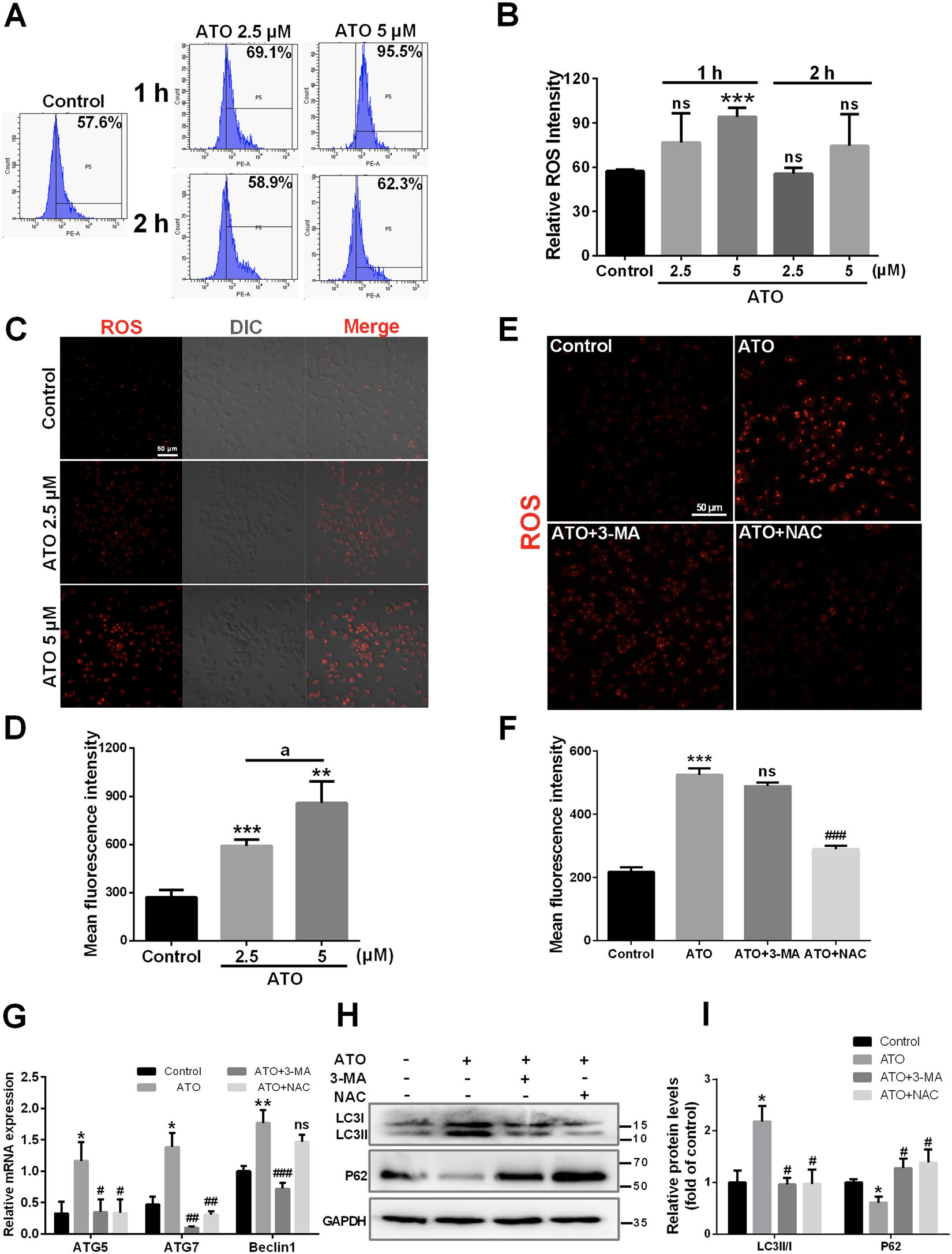
已有研究表明 ROS 促进巨噬细胞中自噬的激活
用ATO诱导巨噬细胞中 ROS 产生的剂量依赖性增加,这种效果在 2 小时时被消除。 此外,NAC 消除了 ROS 的产生,而 3-MA 没有显示出这种效果,说明自噬不会影响ROS产生,ROS不是自噬的下游产物
接下来评估 ROS 是否参与了 ATO 诱导的自噬
NAC与3-MA处理都会抑制 ATO 诱导的自噬相关基因和蛋白质的变化
这些结果表明,增加的 ROS 产生是导致 ATO 诱导巨噬细胞自噬激活的上游事件。
4. ATO刺激巨噬细胞中ROS依赖的TFEB核易位
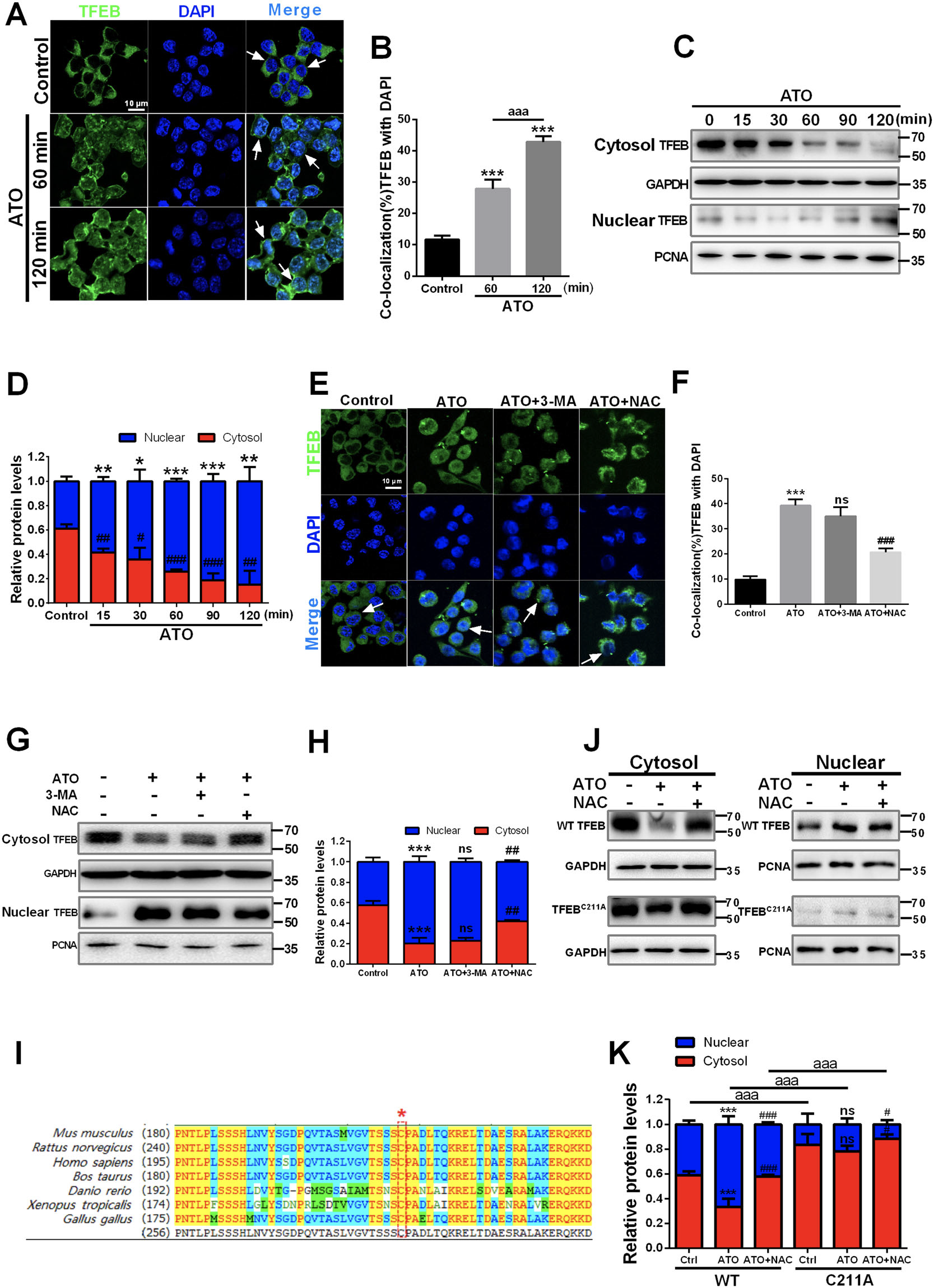
TFEB 是动脉粥样硬化脂质清除的关键调节因子。通常,TFEB 在细胞质中被 mTOR 磷酸化。当去磷酸化时,TFEB 易位到细胞核中以促进自噬体和溶酶体基因表达,从而增强自噬和溶酶体生物发生 。然而,ROS 的产生也可以通过氧化其唯一的半胱氨酸残基来诱导 TFEB 的快速易位
免疫荧光和WB:ATO 处理促进了 TFEB 的核易位和积累,同时降低了其胞质分数。这些效果在120 分钟后更加明显
区分 ROS 生成、自噬激活和 ATO 触发的 TFEB 核易位之间的关系
ATO 处理后TFEB 定位到细胞核中,NAC 可以逆转这一现象,而3-MA不能。表明由 ATO 触发的 TFEB 核易位是 ROS 依赖性的,并且是 ATO 诱导的自噬激活的上游事件。
不同物种TFEB同源物的蛋白序列比对,小鼠 TFEB 中唯一的半胱氨酸残基,半胱氨酸 211,在不同物种中高度保守,推测在 ATO 处理促进这种半胱氨酸残基的氧化是 TFEB 快速核易位的原因。
构建半胱氨酸突变为丙氨酸 (C211A) 的 TFEB 突变体。 在 ATO 处理后,野生型 TFEB 迅速转移到细胞核中。 而TFEB C211A 未能在同一时间内转移到细胞核中。
这些结果表明,ATO 促进了 C211 的氧化,从而导致 TFEB 的快速核转运。
5. ATO促进TFEB依赖的溶酶体生物合成以及巨噬细胞中自噬体和溶酶体的融合
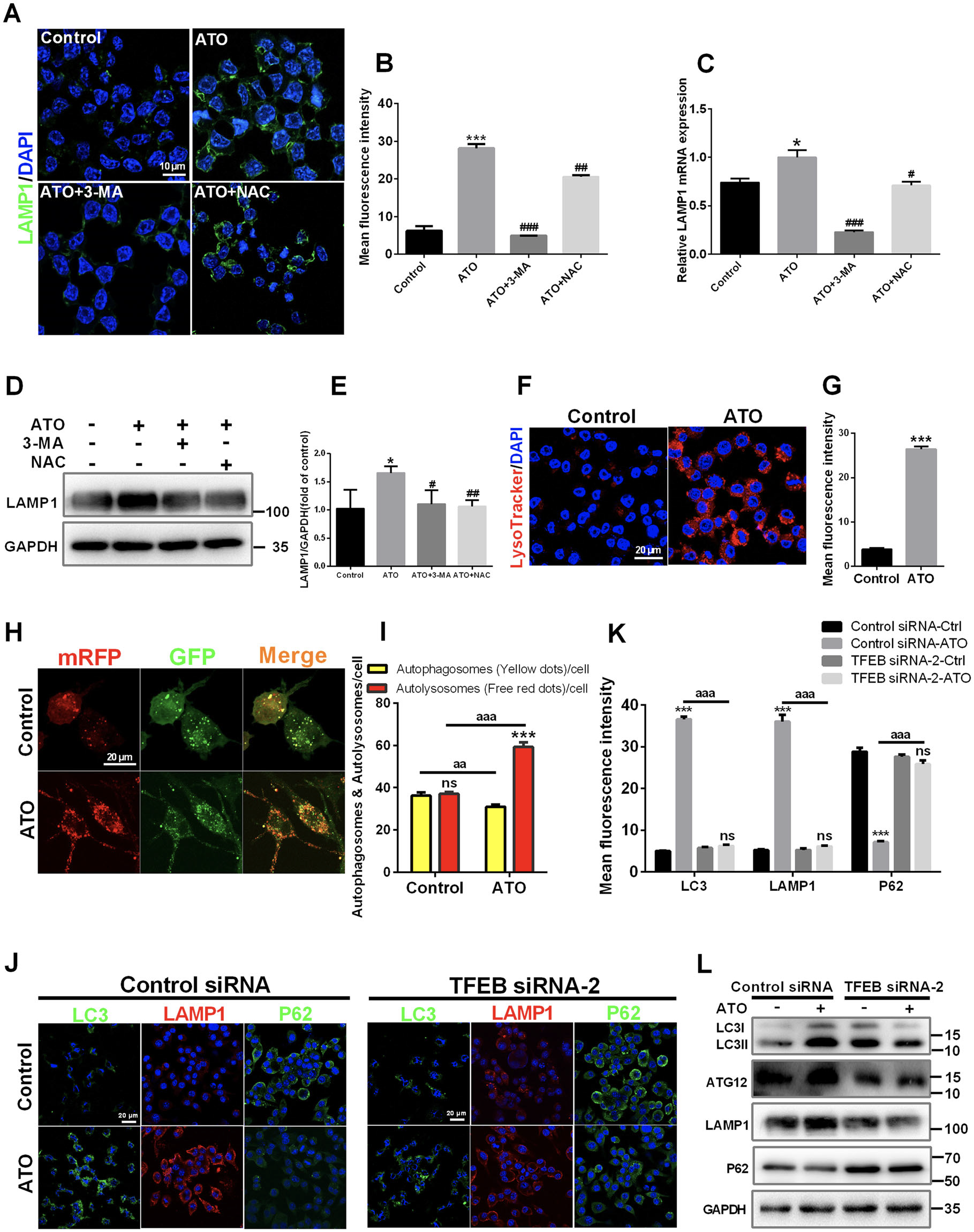
在自噬的最后阶段,自噬体与溶酶体融合并随后降解,这是溶酶体激活所必需的。首先,检查溶酶体生物合成。
ATO增加了巨噬细胞中 LAMP1 的表达。 3-MA 处理消除了 ATO 诱导的 LAMP1 上调,而 NAC 不完全消除了这种现象
在 ATO 处理的巨噬细胞中LysoTracker 染色细胞的荧光增加,表明 ATO 增强了溶酶体酸化
这些结果表明ATO增加了溶酶体的数量并增强了溶酶体的功能,而ROS是这些作用的上游之一。
使用 mRFP-GFP 串联荧光标记 LC3II 来检查自噬体-溶酶体融合。ATO 处理导致 RFP-only 斑点增加,表明 ATO 促进了自噬体-溶酶体融合
TFEB siRNA 用于评估 TFEB 对 ATO 诱导的自噬信号传导的影响,TFEB siRNA后,ATO 诱导LC3II/LC3I、Atg12 和 LAMP1 表达下调,p62表达上调,自噬受到抑制
ATO促进了TFEB依赖性溶酶体的生物合成,以及巨噬细胞中自噬体和溶酶体的融合
6. ATO通过抑制PI3K/AKT/mTOR通路诱导巨噬细胞自噬
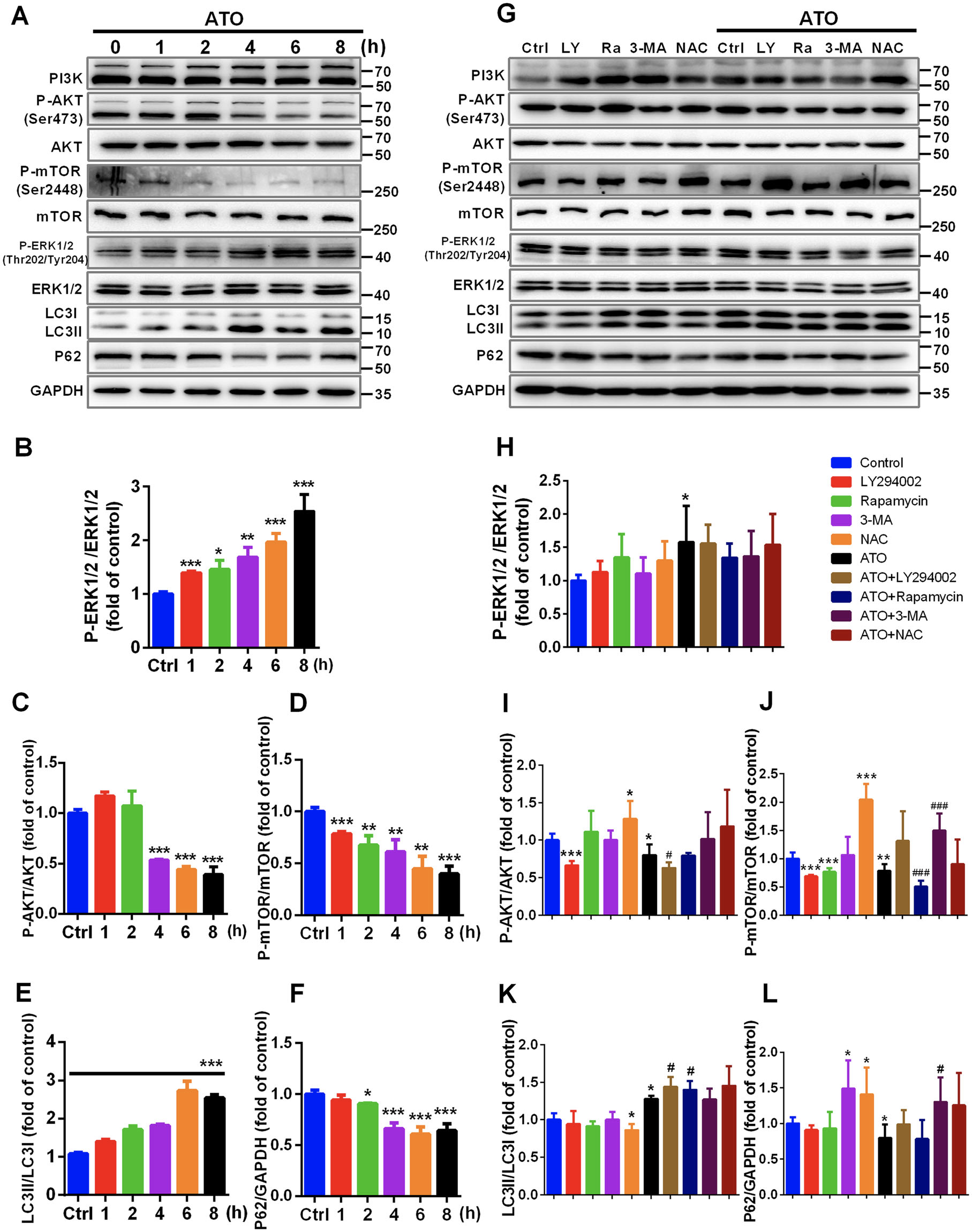
ERK1/2 和 AKT/mTOR 信号级联在调节自噬中起关键作用
为了探索 ATO 对自噬影响的分子机制,分析了ATO 处理不同时间 p-ERK1/2 (Thr202/Tyr204)、ERK1/2、p-AKT (Ser473)、AKT、p-mTOR (Ser2448) 和 mTOR 的水平
表明 PI3K/AKT/mTOR 通路的抑制和pERK1/2的促进是 ATO 诱导自噬的主要特征
为了进一步支持这一结论,巨噬细胞用 I 类 PI3K(LY294002)或 mTOR(雷帕霉素)抑制剂处理。与对照相比,LY294002 抑制 AKT 和 mTOR 磷酸化,而雷帕霉素抑制 mTOR 磷酸化。
LY294002 和雷帕霉素相对于单独使用 ATO 的处理进一步增加了 LC3II/I 比率。 此外,与对照相比,NAC 促进 AKT 和 mTOR 磷酸化,并抑制 LC3II 转化和 p62 降解。NAC 阻止了ATO 诱导的 AKT/mTOR 信号传导以及自噬相关蛋白表达的变化。
这些结果表明 ATO 通过以 ROS 依赖性方式抑制 PI3K/AKT/mTOR 通路来诱导自噬。
7. ATO给药可加速 ApoE−/−小鼠主动脉的自噬,并减少动脉粥样硬化病变
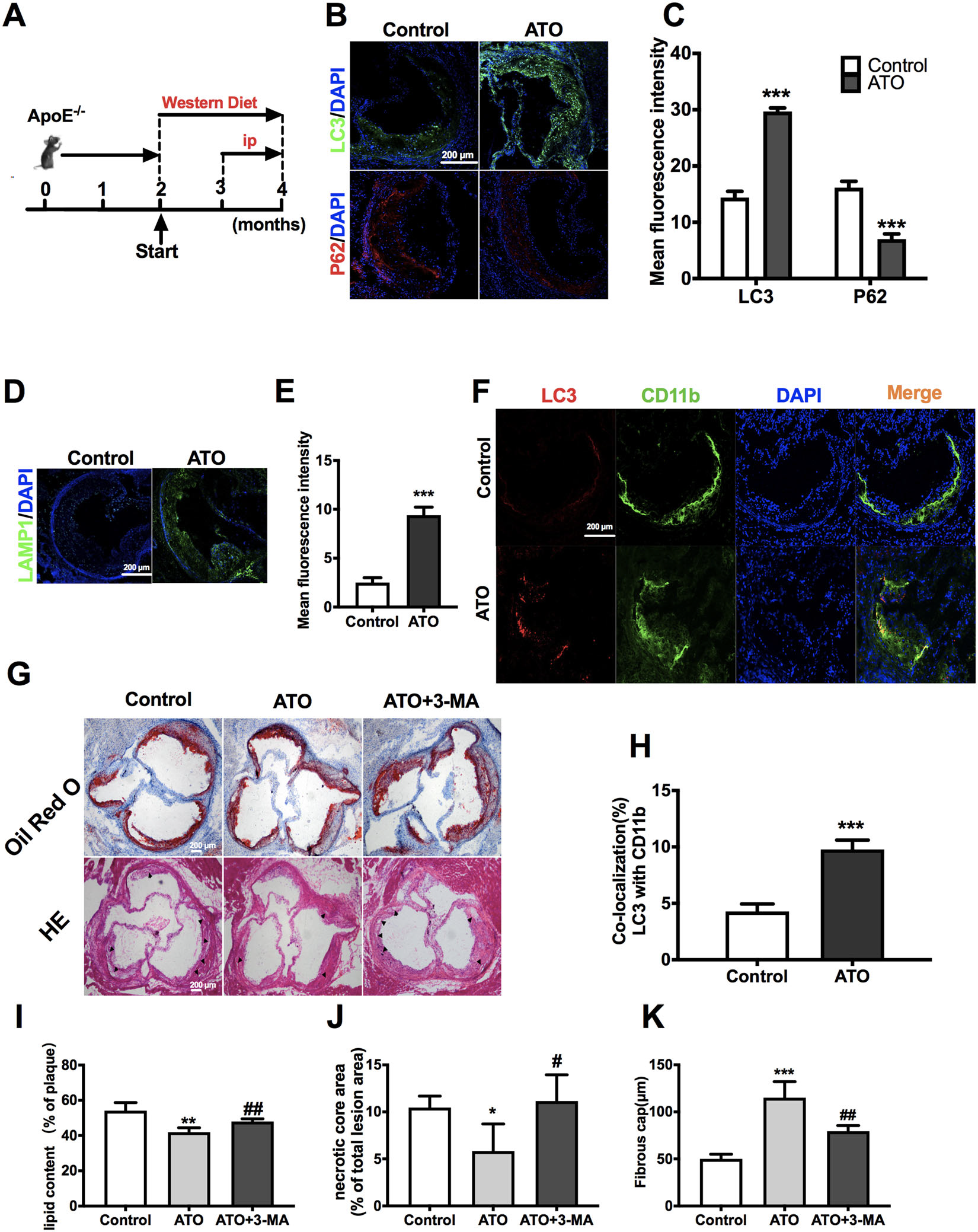
用“西方饮食”喂养 ApoE-/- 小鼠 2 个月以建立早期动脉粥样硬化模型。
从第 12 周到第 16 周,ATO 组每隔一天以 2.5 mg/kg 的剂量腹腔注射 ATO 盐水
免疫荧光:ATO 增加了 LC3 和 LAMP1 的荧光强度,同时降低了主动脉窦中 p62 的荧光强度
ATO 增强了 CD11b+ 细胞中的 LC3 荧光强度。 这些结果表明巨噬细胞在 ATO 诱导的 ApoE-/- 小鼠的主动脉中显示出自噬的特征
为了进一步验证 ATO 诱导的自噬有助于减少动脉粥样硬化病变
将 ATO与 3-MA联合处理,ATO 降低了坏死核心斑块面积和脂质积累的比率,并增加了纤维帽的厚度所有这些 ATO 诱导的变化都被 3-MA 处理逆转
结果共同表明,ATO 通过增强巨噬细胞的自噬减少了早期 AS 的动脉粥样硬化病变。
ATO激活自噬和保护动脉粥样硬化机制的模型

实验技术
背景
动脉粥样硬化是世界上死亡的主要原因。研究证实巨噬细胞凋亡对动脉粥样硬化斑块的发展至关重要。巨噬细胞识别并摄取氧化低密度脂蛋白(ox-LDL),然后转化为泡沫细胞,发生凋亡,最终促进斑块不稳定性。
自噬是一种与凋亡密切相关的自我保护机制,它依赖于溶酶体降解和循环细胞内成分来维持体内稳态。位点自噬流是指自噬的整个动态过程,包括自噬体的形成、自噬体与溶酶体的融合以及自噬体的降解。超过30个自噬相关基因(Atgs)和蛋白参与了自噬过程。LC3I向LC3II的转化是自噬激活的标志,而p62的降解被认为是自噬蛋白降解的标志,p62蛋白的表达量与自噬活性呈现负相关。巨噬细胞自噬通过增加胆固醇流出在动脉粥样硬化中起保护作用。此外,在Ldlr -/-小鼠中,巨噬细胞Atg5缺乏会增加细胞凋亡并促进斑块坏死。因此,基于细胞凋亡和自噬调节的治疗策略可能适合动脉粥样硬化。
自噬的调控是复杂的,涉及多种途径。声动力疗法通过诱导ROS产生促进巨噬细胞自噬。此外,肌磷脂3激酶(PI3K)/蛋白激酶B (AKT)/雷帕霉素(mTOR)是一个经典的自噬途径。同时,激活ERK1/2也可诱导自噬。转录因子EB (TFEB)是mTOR和ERK1/2的下游靶点,是自噬-溶酶体通路的关键调控因子。TFEB上调近三分之二的自噬-溶酶体途径相关基因的表达,其过表达通过挽救脂质诱导的溶酶体功能障碍和下游后遗症,在动脉粥样硬化中具有强大的治疗作用。
诱导自噬和抑制细胞凋亡可能为动脉粥样硬化(AS)提供一种治疗方法,巨噬细胞自噬的调控被证明对动脉粥样硬化非常有益。然而,很少有自噬调节剂的报道。ATO通过促进肿瘤细胞凋亡和自噬死亡在急性早幼粒细胞白血病中发挥治疗作用。提示,ATO可调节巨噬细胞的凋亡和自噬,为动脉粥样硬化的治疗提供了新的视角。
1. 三氧化二砷(ATO)诱导巨噬细胞自噬
MTT实验证明ATO对巨噬细胞和泡沫细胞活力影响具有时间和剂量依赖性
qPCR:ATO处理促进ATG7、ATG5、LC3的表达
WB:ATO增加LC3II/LC3I比值,抑制P62的表达具有时间和剂量依赖性
免疫荧光:表明自噬抑制剂抑制了 ATO诱导的Atg7 mRNA表达、LC3II的积累和p62的降解。
吖啶橙(AO)染色:显示 ATO增加红色信号(酸性细胞器)。另一方面,
3-MA消除了ATO诱导的细胞器酸化
这些数据表明,ATO促进了自噬体的形成和溶酶体的自噬流。
2. 在巨噬细胞和泡沫细胞中,ATO诱导自噬的时间早于抑制凋亡标志物的表达
据报道,自噬和凋亡在不同的阶段被激活。Cleaved caspase-3和Cleaved caspase-9 是可靠的凋亡标志物
为了探讨ATO对自噬和凋亡的动态影响,检测了LC3II、p62、cl-caspase-3和cl-caspase-9的表达
表明 ATO 诱导巨噬细胞2h自噬,抑制细胞凋亡标志物24h、48h的表达
泡沫细胞中 ox-LDL 超载引起的细胞凋亡是一种动脉粥样硬化的迹象。模仿和探索动态ATO在此过程中对自噬和凋亡的影响
结果表明,ATO 2h时诱导泡沫细胞中的自噬,与其在 24 h时对细胞凋亡标志物的抑制相比要早得多。
3. ROS参与了巨噬细胞中ATO诱导的自噬
已有研究表明 ROS 促进巨噬细胞中自噬的激活
用ATO诱导巨噬细胞中 ROS 产生的剂量依赖性增加,这种效果在 2 小时时被消除。 此外,NAC 消除了 ROS 的产生,而 3-MA 没有显示出这种效果,说明自噬不会影响ROS产生,ROS不是自噬的下游产物
接下来评估 ROS 是否参与了 ATO 诱导的自噬
NAC与3-MA处理都会抑制 ATO 诱导的自噬相关基因和蛋白质的变化
这些结果表明,增加的 ROS 产生是导致 ATO 诱导巨噬细胞自噬激活的上游事件。
4. ATO刺激巨噬细胞中ROS依赖的TFEB核易位
TFEB 是动脉粥样硬化脂质清除的关键调节因子。通常,TFEB 在细胞质中被 mTOR 磷酸化。当去磷酸化时,TFEB 易位到细胞核中以促进自噬体和溶酶体基因表达,从而增强自噬和溶酶体生物发生 。然而,ROS 的产生也可以通过氧化其唯一的半胱氨酸残基来诱导 TFEB 的快速易位
免疫荧光和WB:ATO 处理促进了 TFEB 的核易位和积累,同时降低了其胞质分数。这些效果在120 分钟后更加明显
区分 ROS 生成、自噬激活和 ATO 触发的 TFEB 核易位之间的关系
ATO 处理后TFEB 定位到细胞核中,NAC 可以逆转这一现象,而3-MA不能。表明由 ATO 触发的 TFEB 核易位是 ROS 依赖性的,并且是 ATO 诱导的自噬激活的上游事件。
不同物种TFEB同源物的蛋白序列比对,小鼠 TFEB 中唯一的半胱氨酸残基,半胱氨酸 211,在不同物种中高度保守,推测在 ATO 处理促进这种半胱氨酸残基的氧化是 TFEB 快速核易位的原因。
构建半胱氨酸突变为丙氨酸 (C211A) 的 TFEB 突变体。 在 ATO 处理后,野生型 TFEB 迅速转移到细胞核中。 而TFEB C211A 未能在同一时间内转移到细胞核中。
这些结果表明,ATO 促进了 C211 的氧化,从而导致 TFEB 的快速核转运。
5. ATO促进TFEB依赖的溶酶体生物合成以及巨噬细胞中自噬体和溶酶体的融合
在自噬的最后阶段,自噬体与溶酶体融合并随后降解,这是溶酶体激活所必需的。首先,检查溶酶体生物合成。
ATO增加了巨噬细胞中 LAMP1 的表达。 3-MA 处理消除了 ATO 诱导的 LAMP1 上调,而 NAC 不完全消除了这种现象
在 ATO 处理的巨噬细胞中LysoTracker 染色细胞的荧光增加,表明 ATO 增强了溶酶体酸化
这些结果表明ATO增加了溶酶体的数量并增强了溶酶体的功能,而ROS是这些作用的上游之一。
使用 mRFP-GFP 串联荧光标记 LC3II 来检查自噬体-溶酶体融合。ATO 处理导致 RFP-only 斑点增加,表明 ATO 促进了自噬体-溶酶体融合
TFEB siRNA 用于评估 TFEB 对 ATO 诱导的自噬信号传导的影响,TFEB siRNA后,ATO 诱导LC3II/LC3I、Atg12 和 LAMP1 表达下调,p62表达上调,自噬受到抑制
ATO促进了TFEB依赖性溶酶体的生物合成,以及巨噬细胞中自噬体和溶酶体的融合
6. ATO通过抑制PI3K/AKT/mTOR通路诱导巨噬细胞自噬
ERK1/2 和 AKT/mTOR 信号级联在调节自噬中起关键作用
为了探索 ATO 对自噬影响的分子机制,分析了ATO 处理不同时间 p-ERK1/2 (Thr202/Tyr204)、ERK1/2、p-AKT (Ser473)、AKT、p-mTOR (Ser2448) 和 mTOR 的水平
表明 PI3K/AKT/mTOR 通路的抑制和pERK1/2的促进是 ATO 诱导自噬的主要特征
为了进一步支持这一结论,巨噬细胞用 I 类 PI3K(LY294002)或 mTOR(雷帕霉素)抑制剂处理。与对照相比,LY294002 抑制 AKT 和 mTOR 磷酸化,而雷帕霉素抑制 mTOR 磷酸化。
LY294002 和雷帕霉素相对于单独使用 ATO 的处理进一步增加了 LC3II/I 比率。 此外,与对照相比,NAC 促进 AKT 和 mTOR 磷酸化,并抑制 LC3II 转化和 p62 降解。NAC 阻止了ATO 诱导的 AKT/mTOR 信号传导以及自噬相关蛋白表达的变化。
这些结果表明 ATO 通过以 ROS 依赖性方式抑制 PI3K/AKT/mTOR 通路来诱导自噬。
7. ATO给药可加速 ApoE−/−小鼠主动脉的自噬,并减少动脉粥样硬化病变
用“西方饮食”喂养 ApoE-/- 小鼠 2 个月以建立早期动脉粥样硬化模型。
从第 12 周到第 16 周,ATO 组每隔一天以 2.5 mg/kg 的剂量腹腔注射 ATO 盐水
免疫荧光:ATO 增加了 LC3 和 LAMP1 的荧光强度,同时降低了主动脉窦中 p62 的荧光强度
ATO 增强了 CD11b+ 细胞中的 LC3 荧光强度。 这些结果表明巨噬细胞在 ATO 诱导的 ApoE-/- 小鼠的主动脉中显示出自噬的特征
为了进一步验证 ATO 诱导的自噬有助于减少动脉粥样硬化病变
将 ATO与 3-MA联合处理,ATO 降低了坏死核心斑块面积和脂质积累的比率,并增加了纤维帽的厚度所有这些 ATO 诱导的变化都被 3-MA 处理逆转
结果共同表明,ATO 通过增强巨噬细胞的自噬减少了早期 AS 的动脉粥样硬化病变。
ATO激活自噬和保护动脉粥样硬化机制的模型
实验技术

