

坏死性凋亡在肿瘤放疗研究中应用
发布日期:2023-07-20 浏览次数:1281
前言
细胞死亡的主要方式有两种:凋亡(apoptosis)和坏死(necrosis)。凋亡是一种程序化的死亡方式,而坏死是细胞对环境压力的一种被动死亡方式。程序性细胞坏死(Necroptosis)是2005年首次提出的概念,近年来被发现在肿瘤发生和转移中发挥重要作用,并具有治疗肿瘤的潜力。从已发表的文章来看,它已经出现了爆发趋势。
坏死是一种不受调控的细胞死亡形式,由外部物理化学应激而引起。坏死性凋亡则受到高度调控,是细胞面对病毒感染的防御机制。当病毒caspase抑制剂存在时,细胞只能选择以一种不依赖caspase的方式自杀。
坏死性凋亡与凋亡,这两种细胞死亡途径共享着一些上游的信号元件,最终导致质膜破裂,但每个过程的细胞形态有明显差异。坏死性凋亡的特征在于细胞体积增大,细胞器肿胀,细胞膜穿孔,之后是细胞崩解,释放内容物,引发先天和适应性免疫应答,并通过巨胞饮小体清除坏死细胞。凋亡的特征则是细胞皱缩,细胞膜出泡,染色质浓缩,凋亡小体形成,并通过吞噬细胞和巨噬细胞吞噬掉免疫原性的蛋白质。
坏死性凋亡是脊椎动物特有的,源于对病原体的二重防御。当细胞凋亡失效时,坏死性凋亡就成为了“自动防故障机制”。最近的动物研究表明,这种机制能够调节外周组织中的T细胞数量,也是T细胞发育期间清除异常淋巴细胞所必需的。不过,一些疾病也与坏死性凋亡的异常调节有关。
乳腺癌、卵巢癌、黑色素瘤和白血病,这几种癌症都有一个特点,那就是坏死性凋亡的信号蛋白的表达和活性低。相比之下,一些炎性疾病,如多发性硬化症、心肌缺血再灌注损伤、炎症性肠病,则与信号蛋白的表达升高相关联。
Necroptosis的紊乱也是许多炎症性疾病的关键因素
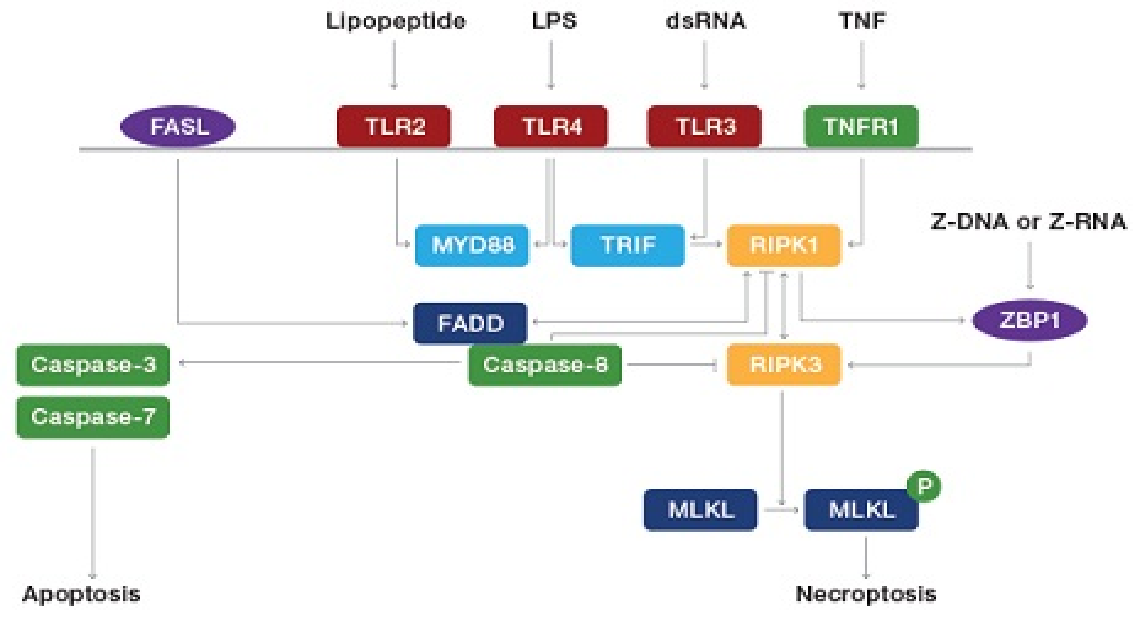
近年来,人们对坏死性凋亡的信号通路进行了广泛研究。它与凋亡有一些共同的上游信号元件,其中研究最深入的是肿瘤坏死因子受体1(TNFR1)。TNFα与质膜上的TNFR1结合,使得TNF受体相关死亡结构域(TRADD)向RIPK1发出信号,招募RIPK3形成坏死体(necrosome)。
caspase-8是坏死性凋亡的关键抑制剂,因为它能裂解和失活RIPK1和RIPK3。如果caspase-8具有活性,那么它与RIPK1和FADD形成复合物,引发细胞凋亡;但如果caspase-8被抑制,那么RIPK1和RIPK3和RIP同型结构域(RHIM)发生相互作用,引发坏死性凋亡。这导致RIPK1和RIPK3之间一系列的自动和交叉磷酸化。
RIPK3的磷酸化导致MLKL的募集和随后的磷酸化,MLKL之后插入并渗透到质膜和细胞器。膜破裂导致细胞内容物溢出进入器官,导致炎症表型的出现和损伤相关分子模式(DAMP)的释放,如IL-1α、IL-β和IL-33等,从而引发免疫应答。DMAP将信号发送到循环系统,将免疫细胞招募到受损的组织。巨噬细胞中的组分巨胞饮小体(macropinosome)通过胞饮作用清除坏死性凋亡细胞。
文献案例
Necroptosis regulates tumor repopulation after radiotherapy via RIP1/RIP3/MLKL/JNK/IL8 pathway
坏死性凋亡通过 RIP1/RIP3/MLKL/JNK/IL8 通路调节放疗后肿瘤再增殖
期刊:J Exp Clin Cancer Res.
发表时间:2019年11月
影响因子:11.1613
研究背景
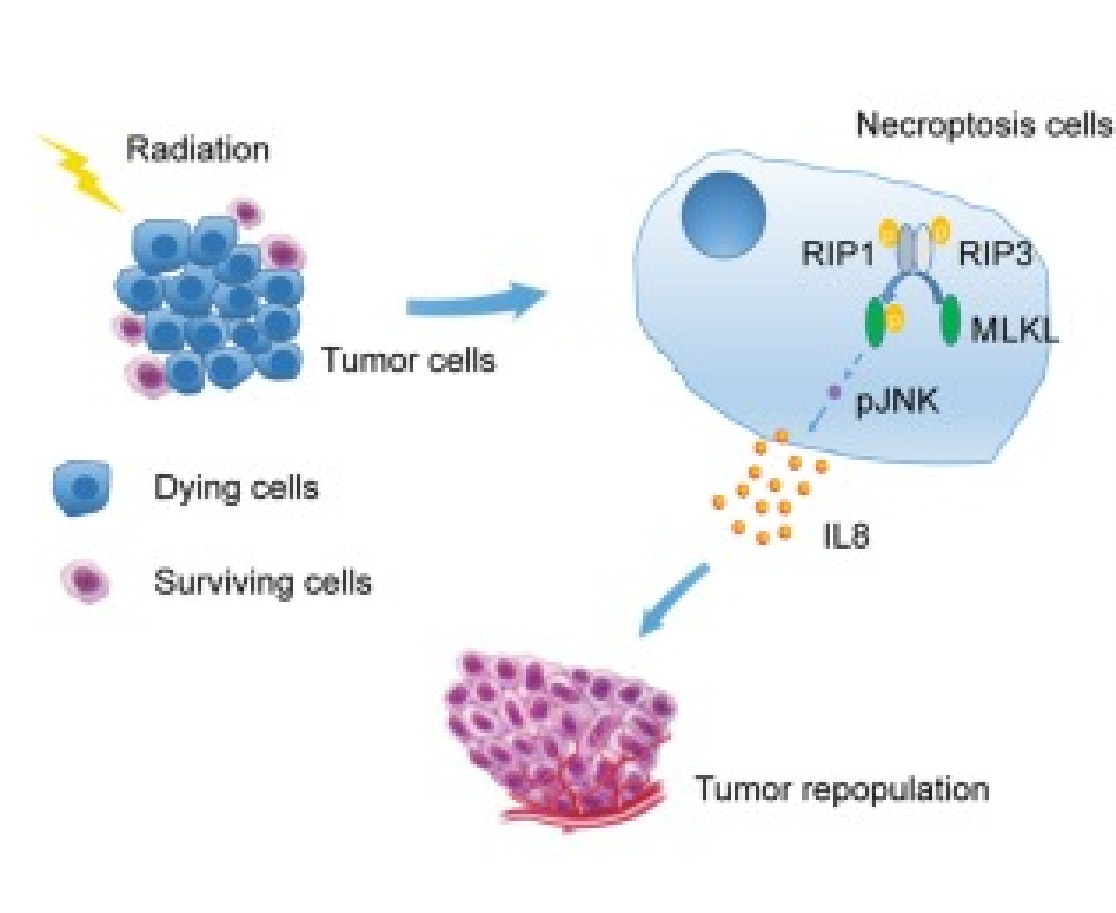
放射治疗(RT)是癌症的主要治疗方式之一。超过50%的癌症患者在疾病治疗过程中接受了放疗,其中40%可以通过放疗治愈。然而,肿瘤再增殖仍然是治疗失败的关键因素之一 。肿瘤再增殖是一些存活的肿瘤细胞在放射治疗期间或之后甚至加速增殖的过程。许多研究致力于探索这一过程的分子机制,据报道,一些与生长相关的信号通路、癌症干细胞和肿瘤微环境(如缺氧、受肿瘤教育的巨噬细胞或成纤维细胞)与此有关。
细胞凋亡和坏死已被认为是癌症治疗的积极过程。然而,之前的研究表明,细胞凋亡和坏死可能通过激活增殖信号通路并在细胞死亡期间产生生长因子来促进肿瘤生长。例如,照射后与凋亡相关的 Caspase-3 的激活导致钙非依赖性磷脂酶 A2 (iPLA2) 裂解和活化,从而增加了花生四烯酸 (AA) 的合成以及随后的前列腺素 E2 (PGE2) 的产生和释放。凋亡肿瘤细胞释放的 PGE2 然后刺激存活肿瘤细胞的增殖以及血管生成。还发现,受辐射的坏死肿瘤细胞释放的高迁移率组框 1 (HMGB1) 参与了肿瘤的再增殖 。
除了细胞凋亡外,一种称为坏死性凋亡的新型可编程形式的坏死最近引起了人们的关注。RIP3和MLKL的激活都被认为是坏死性凋亡的生物标志物。TNF/TNF 受体 1 介导的信号通路是研究最广泛的坏死性凋亡模型之一,广泛存在于不同类型的肿瘤和其他病理生理状况中。在 TNF/TNFR1 诱导的坏死期间,RIP1 首先通过磷酸化被激活,进而通过其激酶活性激活 RIP3,它们一起形成 RIP1/RIP3 复合物,这导致 MLKL 的磷酸化。磷酸化的 MLKL 被认为是 TNF 驱动的坏死性凋亡的生物标志物,然后被转运到细胞核和细胞膜,最终引发细胞膜破裂和细胞死亡。因此,RIP1 的化学抑制剂,例如 Necrostatin-1 (Nec-1),可以特别抑制 TNF 驱动的坏死性凋亡 。据报道,内分泌癌中辐射诱导的细胞死亡存在坏死性凋亡。然而,坏死性凋亡在放射相关癌症治疗中的作用尚不清楚。特别是,这种新的细胞死亡形式是否以及如何参与与辐射相关的肿瘤再增殖尚不清楚。
放疗后肿瘤细胞再增殖是肿瘤放疗抵抗和复发的主要原因。本研究旨在探讨放疗后肿瘤再增殖的潜在机制,重点关注坏死性凋亡是否以及如何参与这一过程。
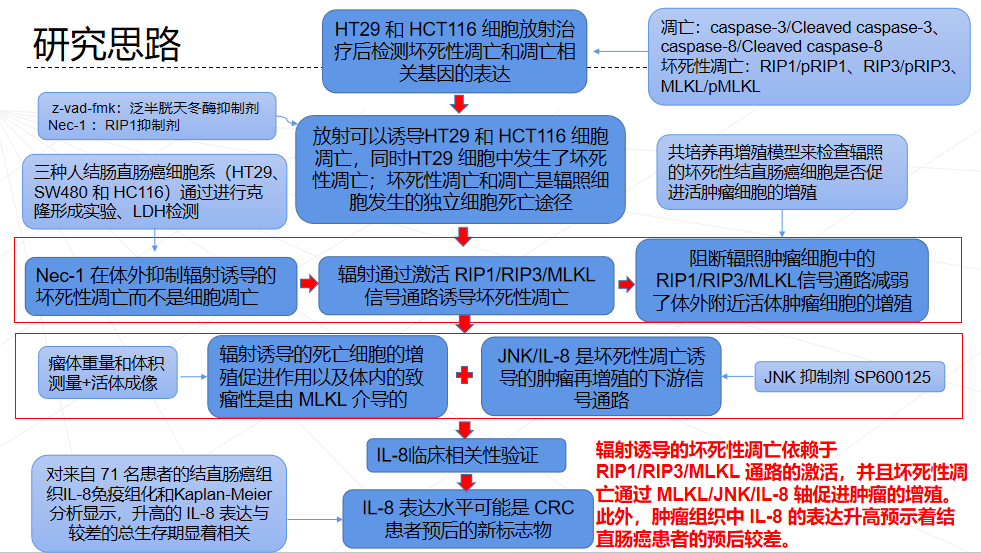
相关指标监测:
1. 凋亡相关检测指标:caspase-3/Cleaved caspase-3、caspase-8/Cleaved caspase-8
2. 坏死性凋亡相关检测指标:RIP1/pRIP1、RIP3/pRIP3、MLKL/pMLKL
3. 乳酸脱氢酶(LDH)
相关抑制剂
1. z-vad-fmk:泛半胱天冬酶抑制剂
2. Nec-1:RIP1 抑制剂
3. GSK'872: RIP3 抑制剂
4. Necrosulfonamide:MLKL 抑制剂
创新点
1. 首次证明了坏死性凋亡参与与辐射相关的肿瘤再增殖
2. 阐明了辐射诱导的坏死性凋亡依赖于 RIP1/RIP3/MLKL 通路的激活,并且坏死性凋亡通过 MLKL/JNK/IL-8 轴促进肿瘤的增殖
前言
细胞死亡的主要方式有两种:凋亡(apoptosis)和坏死(necrosis)。凋亡是一种程序化的死亡方式,而坏死是细胞对环境压力的一种被动死亡方式。程序性细胞坏死(Necroptosis)是2005年首次提出的概念,近年来被发现在肿瘤发生和转移中发挥重要作用,并具有治疗肿瘤的潜力。从已发表的文章来看,它已经出现了爆发趋势。
坏死是一种不受调控的细胞死亡形式,由外部物理化学应激而引起。坏死性凋亡则受到高度调控,是细胞面对病毒感染的防御机制。当病毒caspase抑制剂存在时,细胞只能选择以一种不依赖caspase的方式自杀。
坏死性凋亡与凋亡,这两种细胞死亡途径共享着一些上游的信号元件,最终导致质膜破裂,但每个过程的细胞形态有明显差异。坏死性凋亡的特征在于细胞体积增大,细胞器肿胀,细胞膜穿孔,之后是细胞崩解,释放内容物,引发先天和适应性免疫应答,并通过巨胞饮小体清除坏死细胞。凋亡的特征则是细胞皱缩,细胞膜出泡,染色质浓缩,凋亡小体形成,并通过吞噬细胞和巨噬细胞吞噬掉免疫原性的蛋白质。
坏死性凋亡是脊椎动物特有的,源于对病原体的二重防御。当细胞凋亡失效时,坏死性凋亡就成为了“自动防故障机制”。最近的动物研究表明,这种机制能够调节外周组织中的T细胞数量,也是T细胞发育期间清除异常淋巴细胞所必需的。不过,一些疾病也与坏死性凋亡的异常调节有关。
乳腺癌、卵巢癌、黑色素瘤和白血病,这几种癌症都有一个特点,那就是坏死性凋亡的信号蛋白的表达和活性低。相比之下,一些炎性疾病,如多发性硬化症、心肌缺血再灌注损伤、炎症性肠病,则与信号蛋白的表达升高相关联。
Necroptosis的紊乱也是许多炎症性疾病的关键因素
近年来,人们对坏死性凋亡的信号通路进行了广泛研究。它与凋亡有一些共同的上游信号元件,其中研究最深入的是肿瘤坏死因子受体1(TNFR1)。TNFα与质膜上的TNFR1结合,使得TNF受体相关死亡结构域(TRADD)向RIPK1发出信号,招募RIPK3形成坏死体(necrosome)。
caspase-8是坏死性凋亡的关键抑制剂,因为它能裂解和失活RIPK1和RIPK3。如果caspase-8具有活性,那么它与RIPK1和FADD形成复合物,引发细胞凋亡;但如果caspase-8被抑制,那么RIPK1和RIPK3和RIP同型结构域(RHIM)发生相互作用,引发坏死性凋亡。这导致RIPK1和RIPK3之间一系列的自动和交叉磷酸化。
RIPK3的磷酸化导致MLKL的募集和随后的磷酸化,MLKL之后插入并渗透到质膜和细胞器。膜破裂导致细胞内容物溢出进入器官,导致炎症表型的出现和损伤相关分子模式(DAMP)的释放,如IL-1α、IL-β和IL-33等,从而引发免疫应答。DMAP将信号发送到循环系统,将免疫细胞招募到受损的组织。巨噬细胞中的组分巨胞饮小体(macropinosome)通过胞饮作用清除坏死性凋亡细胞。
文献案例
Necroptosis regulates tumor repopulation after radiotherapy via RIP1/RIP3/MLKL/JNK/IL8 pathway
坏死性凋亡通过 RIP1/RIP3/MLKL/JNK/IL8 通路调节放疗后肿瘤再增殖
期刊:J Exp Clin Cancer Res.
发表时间:2019年11月
影响因子:11.1613
研究背景
放射治疗(RT)是癌症的主要治疗方式之一。超过50%的癌症患者在疾病治疗过程中接受了放疗,其中40%可以通过放疗治愈。然而,肿瘤再增殖仍然是治疗失败的关键因素之一 。肿瘤再增殖是一些存活的肿瘤细胞在放射治疗期间或之后甚至加速增殖的过程。许多研究致力于探索这一过程的分子机制,据报道,一些与生长相关的信号通路、癌症干细胞和肿瘤微环境(如缺氧、受肿瘤教育的巨噬细胞或成纤维细胞)与此有关。
细胞凋亡和坏死已被认为是癌症治疗的积极过程。然而,之前的研究表明,细胞凋亡和坏死可能通过激活增殖信号通路并在细胞死亡期间产生生长因子来促进肿瘤生长。例如,照射后与凋亡相关的 Caspase-3 的激活导致钙非依赖性磷脂酶 A2 (iPLA2) 裂解和活化,从而增加了花生四烯酸 (AA) 的合成以及随后的前列腺素 E2 (PGE2) 的产生和释放。凋亡肿瘤细胞释放的 PGE2 然后刺激存活肿瘤细胞的增殖以及血管生成。还发现,受辐射的坏死肿瘤细胞释放的高迁移率组框 1 (HMGB1) 参与了肿瘤的再增殖 。
除了细胞凋亡外,一种称为坏死性凋亡的新型可编程形式的坏死最近引起了人们的关注。RIP3和MLKL的激活都被认为是坏死性凋亡的生物标志物。TNF/TNF 受体 1 介导的信号通路是研究最广泛的坏死性凋亡模型之一,广泛存在于不同类型的肿瘤和其他病理生理状况中。在 TNF/TNFR1 诱导的坏死期间,RIP1 首先通过磷酸化被激活,进而通过其激酶活性激活 RIP3,它们一起形成 RIP1/RIP3 复合物,这导致 MLKL 的磷酸化。磷酸化的 MLKL 被认为是 TNF 驱动的坏死性凋亡的生物标志物,然后被转运到细胞核和细胞膜,最终引发细胞膜破裂和细胞死亡。因此,RIP1 的化学抑制剂,例如 Necrostatin-1 (Nec-1),可以特别抑制 TNF 驱动的坏死性凋亡 。据报道,内分泌癌中辐射诱导的细胞死亡存在坏死性凋亡。然而,坏死性凋亡在放射相关癌症治疗中的作用尚不清楚。特别是,这种新的细胞死亡形式是否以及如何参与与辐射相关的肿瘤再增殖尚不清楚。
放疗后肿瘤细胞再增殖是肿瘤放疗抵抗和复发的主要原因。本研究旨在探讨放疗后肿瘤再增殖的潜在机制,重点关注坏死性凋亡是否以及如何参与这一过程。
相关指标监测:
1. 凋亡相关检测指标:caspase-3/Cleaved caspase-3、caspase-8/Cleaved caspase-8
2. 坏死性凋亡相关检测指标:RIP1/pRIP1、RIP3/pRIP3、MLKL/pMLKL
3. 乳酸脱氢酶(LDH)
相关抑制剂
1. z-vad-fmk:泛半胱天冬酶抑制剂
2. Nec-1:RIP1 抑制剂
3. GSK'872: RIP3 抑制剂
4. Necrosulfonamide:MLKL 抑制剂
创新点
1. 首次证明了坏死性凋亡参与与辐射相关的肿瘤再增殖
2. 阐明了辐射诱导的坏死性凋亡依赖于 RIP1/RIP3/MLKL 通路的激活,并且坏死性凋亡通过 MLKL/JNK/IL-8 轴促进肿瘤的增殖
- 上一篇:巨噬细胞分型与肿瘤微环境
- 下一篇:骨质疏松机制研究

